


Sequence Comparison
NB: Be careful when using these programmes; it is possible to
align one sequence with any other, if you really want to. False
alignments, and the research you plan using them, may have no biological significance!
We will begin with the most common use of bestfit - to find the best
region of similarity between two distantly-related (but homologous) sequences.
prompt> fetch gb_in:pdrhod -out=pdrhod.gb_in
prompt> gapshow pdrhod.ge_in rnops.ge_ro -begin1=982
begin2=961 end1=1022 end2=1001

Comparing Two Sequences
There are three variations on the theme of sequence comparison. You can find
the BEST region of similarity between two sequences, the best OVERALL
alignment of two sequences, or ALL regions of similarity between them.
E/GCG provides three programme sets for these three different tasks:
prompt> fetch gb_ro:rnops -out=rnops.gb_ro
prompt> bestfit pdrhod.gb_in rnops.gb_ro -out=rhodop.pair
BestFit makes an optimal alignment of the best segment of similarity
between two sequences. Optimal alignments are found by inserting gaps to
maximize the number of matches using the local homology algorithm of
Smith and Waterman.
Begin (* 1 *) ?
End (* 1675 *) ?
Reverse (* No *) ?
Begin (* 1 *) ?
End (* 1493 *) ?
Reverse (* No *) ?
What is the gap creation penalty (* 5.00 *) ?
What is the gap extension penalty (* 0.30 *) ?
Aligning ..................................................
........................-.
Gaps: 0
Quality: 20.1
Quality Ratio: 0.490
% Similarity: 73.171
Length: 41
prompt>
prompt> more rhodop.pair
BESTFIT of: pdrhod.ge_in check: 8638 from: 1 to: 1675
LOCUS PDRHOD 1675 bp RNA INV 12-SEP-1993
DEFINITION Octopus mRNA for rhodopsin.
ACCESSION X07797
NID g9822
KEYWORDS rhodopsin.
SOURCE Octopus dofleini. . . .
to: rnops.ge_ro check: 6230 from: 1 to: 1493
LOCUS RNOPS 1493 bp RNA ROD 20-DEC-1994
DEFINITION R.norvegicus mRNA for rhodopsin.
ACCESSION Z46957
NID g603874
KEYWORDS rhodopsin.
SOURCE Norway rat. . . .
Symbol comparison table: /usr/prog/gcg/gcgcore/data/rundata/swgapdna.cmp
CompCheck: 5234
Gap Weight: 5.000 Average Match: 1.000
Length Weight: 0.300 Average Mismatch: -0.900
Quality: 20.1 Length: 41
Ratio: 0.490 Gaps: 0
Percent Similarity: 73.171 Percent Identity: 73.171
pdrhod.ge_in x rnops.ge_ro September 24, 1996 10:00 ..
. . . .
982 TGTTTGCTAAAGCTTCAGCTATCCACAACCCAATTGTCTAC 1022
| |||||||| | | | ||| |||||||||| |||||
961 TCTTTGCTAAGACCGCCTCCATCTACAACCCAATCATCTAC 1001
prompt>
On-line help for bestfit and gapshow
is available via the commands
&
gapshow.
prompt> genhelp bestfit
You may also check the manual web pages for complete details:
bestfit
prompt> genhelp gapshow
gap is for aligning two sequences over their entire length. While it will work with distantly-related sequences (as in the example above), much of the alignment may have little to no biological significance. Instead, we will align two more closely-related rhodopsin mRNAs.
prompt>lookup -library=genbank -definition=rhodopsin
-organism="Oryctolagus cuniculus" -out=rabbitrh.list
prompt> more rabbitrh.list
LOOKUP in: genbank of: "([SQ-DEF: rhodopsin*] & [SQ-ORG: oryctolagus cuniculus*])"
1 entry September 24, 1996 14:52 ..
gb:OCU21688 ! ID: a4f20006
! DEFINITION Oryctolagus cuniculus rhodopsin mRNA, complete cds.
prompt> fetch gb:OCU21688 -out=ocops.gb_om
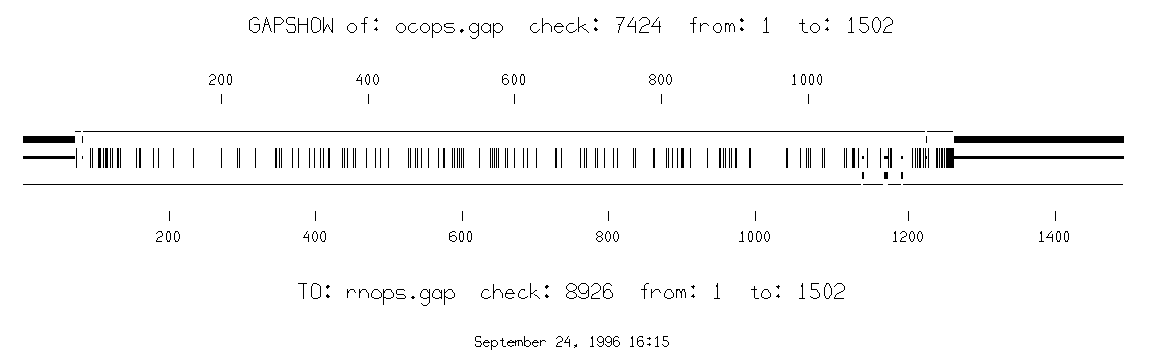
prompt> gap ocops.gb_om rnops.gb_ro -out=rhodopm.pair -outfile2=ocops.gap -outfile3=rnops.gap
Gap uses the algorithm of Needleman and Wunsch to find the alignment of
two complete sequences that maximizes the number of matches and minimizes
the number of gaps.
Begin (* 1 *) ?
End (* 1198 *) ?
Reverse (* No *) ?
Begin (* 1 *) ?
End (* 1493 *) ?
Reverse (* No *) ?
What is the gap creation penalty (* 5.00 *) ?
What is the gap extension penalty (* 0.30 *) ?
Aligning ..................................................
.........-......
Gaps: 5
Quality: 1004.7
Quality Ratio: 0.839
% Similarity: 86.880
Length: 1502
prompt>
prompt> more rhodopm.pair
prompt>
gapshow ocops.gap
rnops.gap
compare, together with the graphing programme dotplot, is used to show regions of similarity within a sequence or between two sequences.
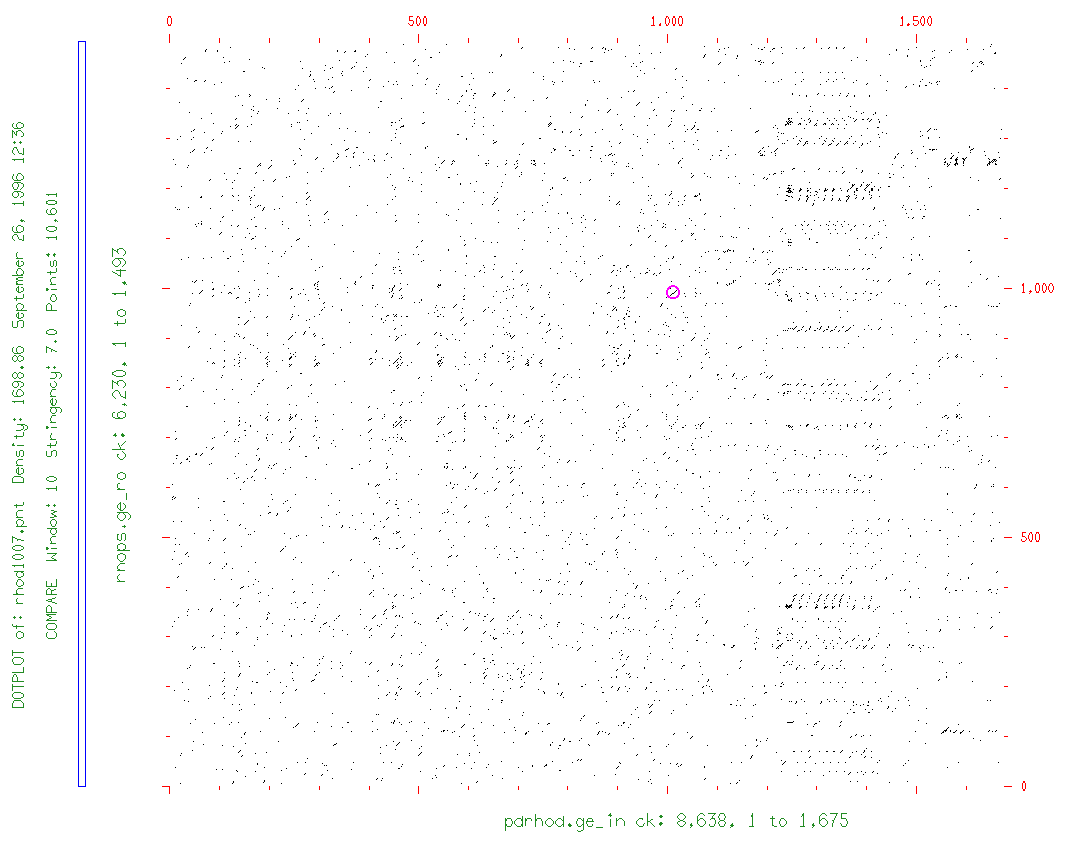
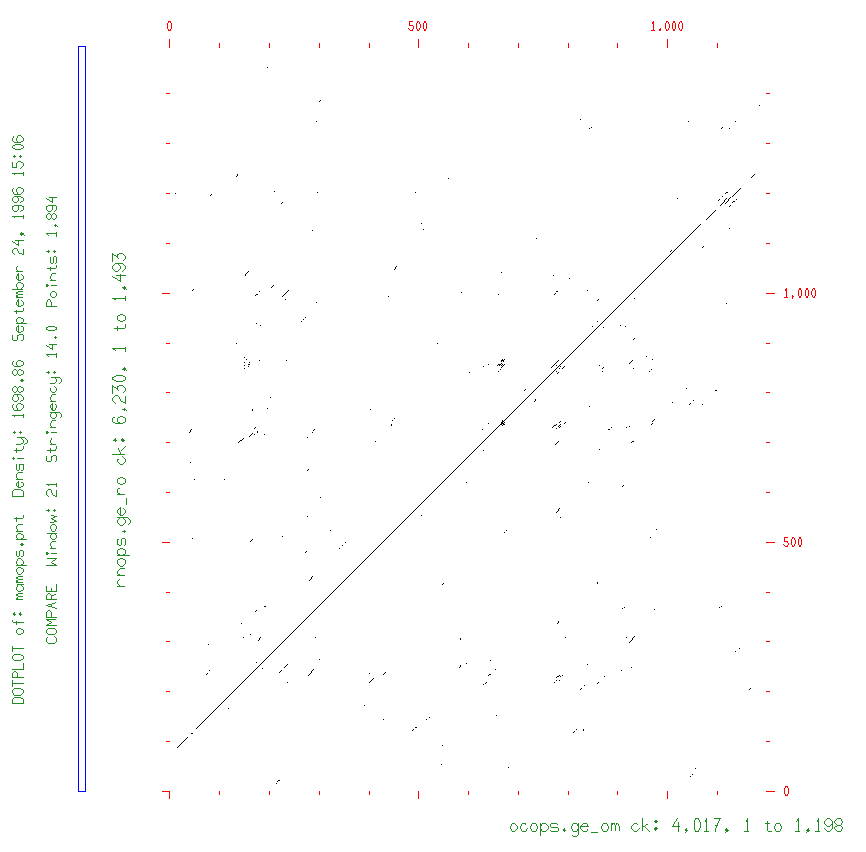
In the example sequences for bestfit, the two distantly-related rhodopsin mRNAs showed a best alignment region having ~73% similarity, and a second best one with ~70%; overall, though, these two sequences have only ~43% similarity (data from gap not shown). Thus, for compare to show only the best regions of similarity for the two distantly-related sequences, we need to use a stringency of between 60% & 70% matching bases. When compare checks for the percentage of matching bases, it does so in every possible comparison register, and within a window, i.e., a certain number of bases at a time. In a window of size 10, at least 6 to 7 bases must match (our best alignment region stringency conditions) for compare to score a "hit" between the two sequences.
prompt> compare pdrhod.ge_in rnops.ge_ro
...
prompt> dotplot pdrhod.pnt
prompt> compare pdrhod.ge_in rnops.ge_ro -win=41
-stri=28 -out=rhod4128.pnt
...
prompt> dotplot rhod4128.pnt
Generally, distantly-related sequences reveal their significant homologies when the window size is high and the stringency is low. With closely-related sequences, a medium size window with high stringency is best. E/GCG recommends the default window size of 21 and stringency of 67% (14) only as a starting point.
prompt> compare pdrhod.ge_in rnops.ge_ro -win=100
-stri=40 -out=rhod0040.pnt
...
prompt> dotplot rhod0040.pnt
prompt> compare ocops.ge_om rnops.ge_ro
-out=rhodm2114.pnt
...
prompt> dotplot rhodm2114.pnt
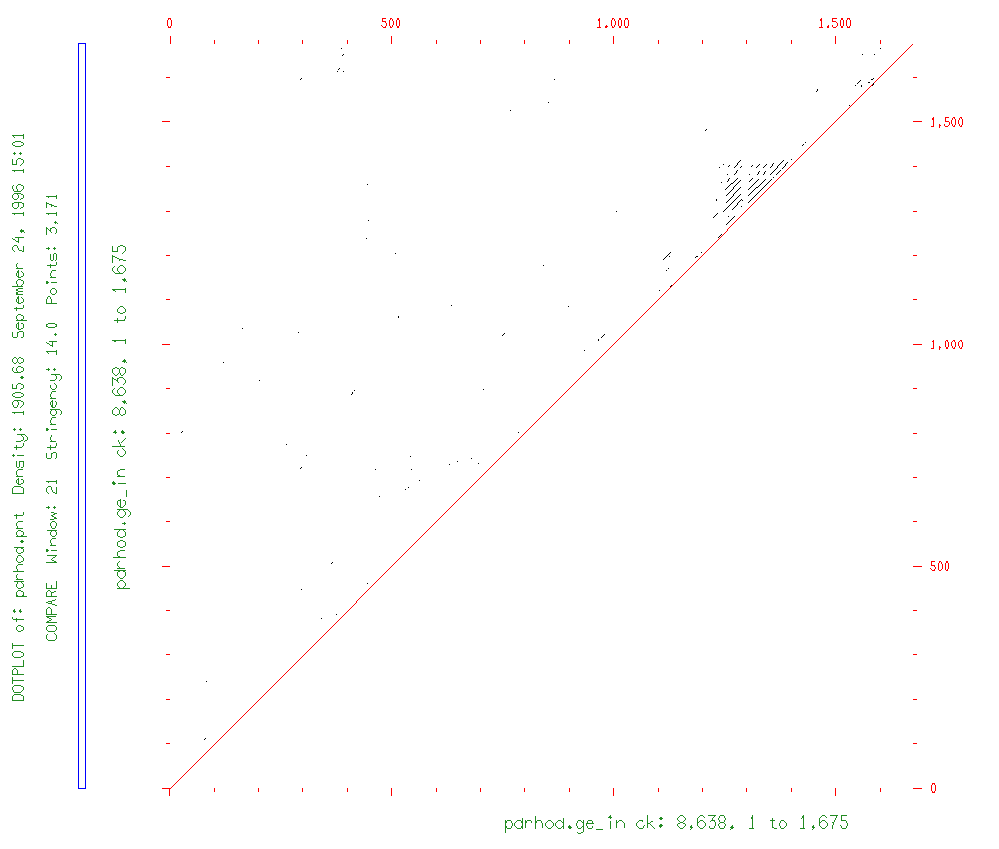
prompt> compare pdrhod.ge_in pdrhod.ge_in
-out=pdrhod2114.pnt
...
prompt> dotplot pdrhod2114.pnt -all
Please continue with Part
8 - Searching Databases - 1 


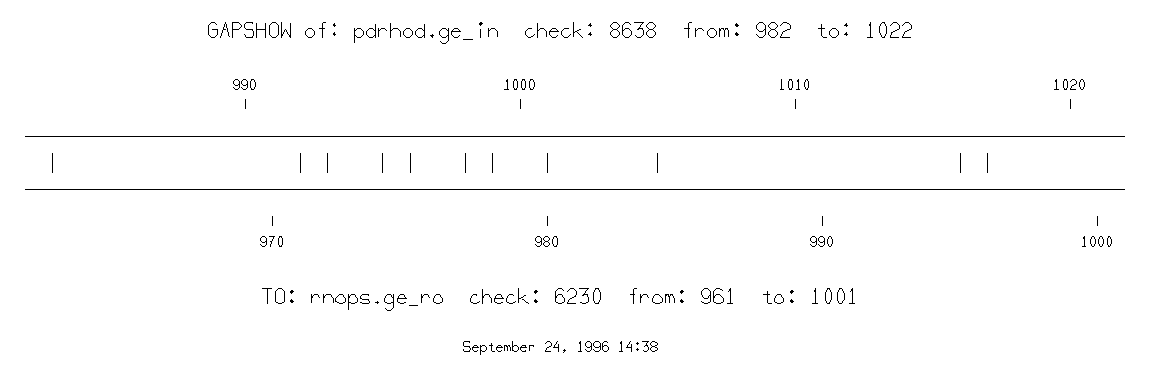{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}